DO YOUR MUSCLES LOCK UP?
Do the muscles in your hands, legs or eyelids sometimes “get stuck” when used? It could be a sign of NDM.
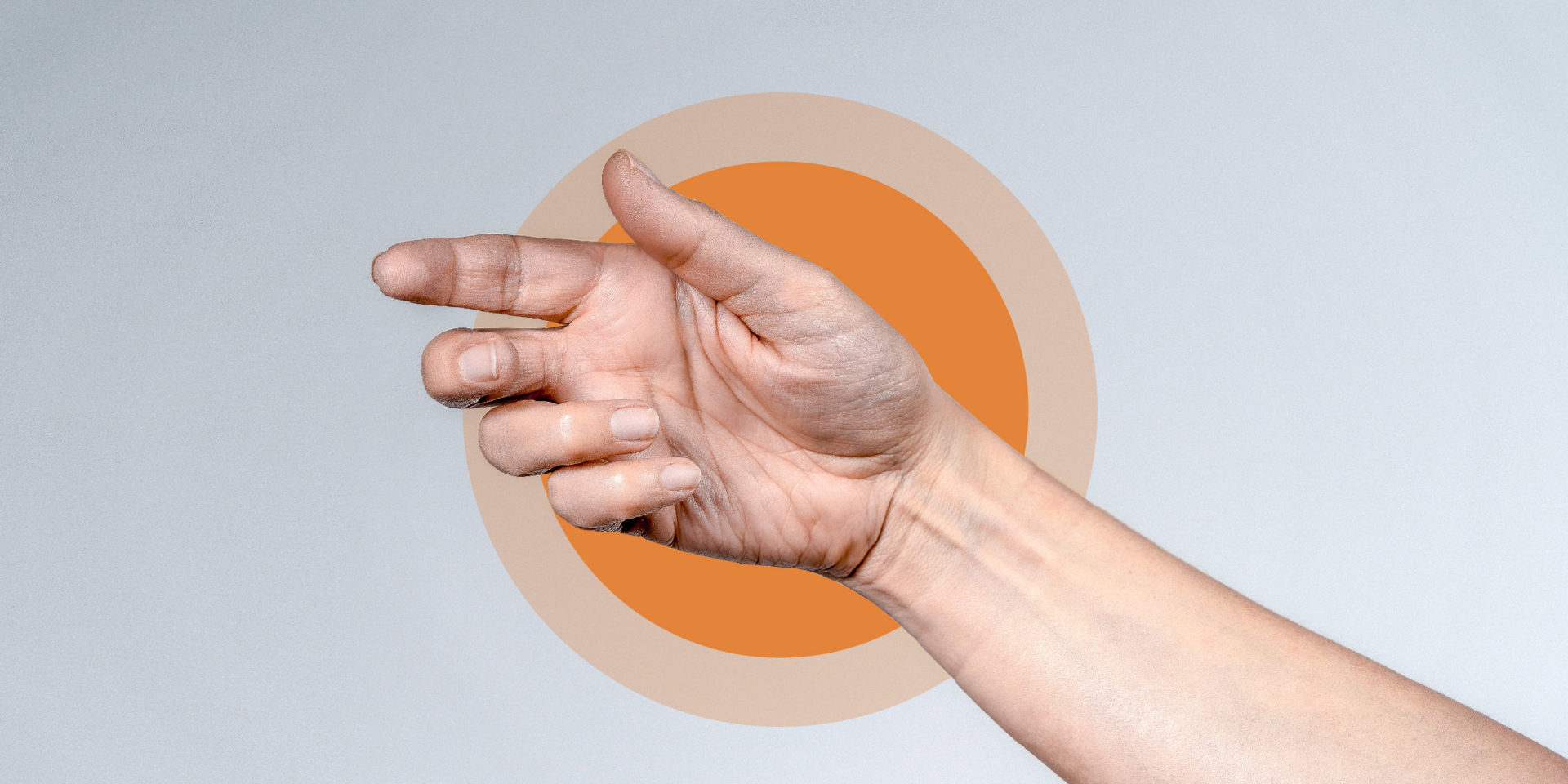
An awareness campaign by Lupin Healthcare Ltd designed for the public and patients with NDM
I'm worried about my symptoms
Learn about non-dystrophic myotonia, its causes and treatment.
What is NDM?
NDM is a group of rare conditions that affects one’s muscles. Only around 1 in 100,000 people worldwide suffer from these conditions, but there are countries where NDM is more common.1 There are several different forms of NDM, with 4 main types commonly known as Becker Myotonia Congenita, Thomsen Myotonia Congenita, Paramyotonia Congenita (Eulenburg) and Hyperkalemic periodic paralysis (hyperPP).1,2
NDM disorders typically affect the muscles, with patients most commonly having an inability to relax muscles immediately after they have been used, which is called myotonia.1 Patients experience myotonia symptoms differently, describing their experience as stiffness, cramps or “locking” of their muscles during everyday tasks and movements.3 In addition to patients experiencing their symptoms differently, patients often describe their symptoms as unpredictable as they may differ from time to time4,5 and they may range from subtle effects on muscles that have little impact on the ability to move to more severe symptoms that significantly affect daily activities.1,3
Struggling to open your hand after clenching your fist may be a sign that you have NDM.6
The voices of people living with NDM








References
- Hahn C, Salajegheh MK. Iran J Neurol 2016; 15(1):46–53
- Trivedi JR, et al. Brain 2013;136:2189–2200
- Nowak U, et al. Presented at WMS20: Virtual Congress. 28 Sep–20 Oct 2020
- Heatwole CR, et al. Muscle Nerve 2013;47:632–48
- Trip J, et al. J Neurol 2009;256:939–47
- Heatwole CR, et al. ASENT 2007 Apr;4:238-251
UK-NDM-2405-00001 May 2024